
Research objects
We work with the proteins which form amyloid fibrils or droplets.
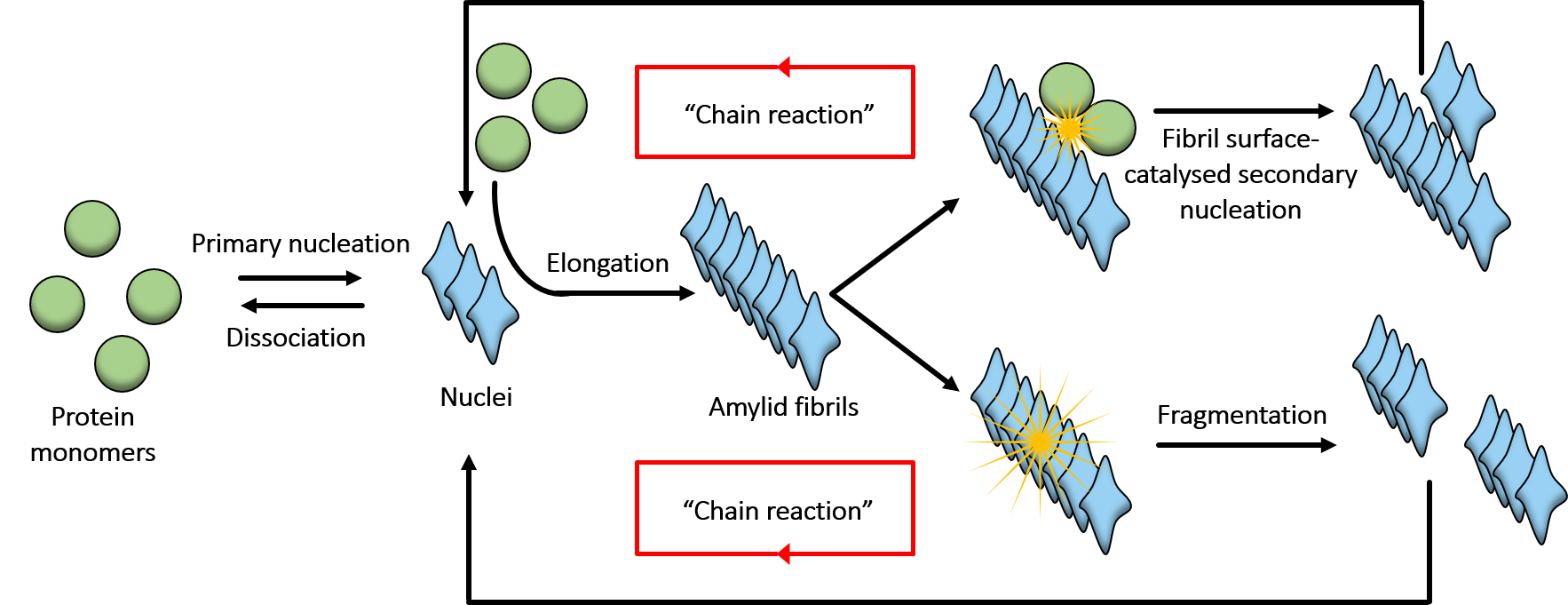
- Amyloid fibrils are protein aggregates with cross-beta sheet structures. They are found during neurodegenerative disorders, cardiac diseases and cancer.
- Protein droplets form during liquid-liquid phase separation, which can happen inside the cell. LLPS initiate protein aggregation, but it is also associated with several protein functions.
In our laboratory, we work with these proteins:
- Alzheimer's disease:beta-amyloid, S100A, Tau; Tau baltymas.
- Parkinson's disease:alpha-synuclein;
- Spongiform encephalopathies: prion proteins;
- Amyotrophic lateral sclerosis:Superoxide dismutase 1 (SOD1);
- Other amyloidoses:ß2-microglobulin, transthyretin;
- Model proteins: insulin, lysozyme;
- Bacterial and yeast proteins:CsgABC, Sup35, Ure2;
- Chaperones and regulatory proteins:14-3-3, Hsp, DegP;
Methodology
- Gene engineering;
- Protein and nucleic acid electrophoresis;
- Production of recombinant proteins in bacteria;
- Chromatographic methods (e.g. ion exchange chromatography, affinity chromatography, size exclusion chromatography, high-performance liquid chromatography (HPLC)) to purify proteins and other molecules of interest;
- Monitoring of the amyloid aggregation process by measuring the absorption or dynamic light scattering of the solution, or the intensity of amyloid-specific dye fluorescence emission. Subsequently, we analyse aggregation kinetics using chemical kinetics tools;
- Infrared spectroscopy to obtain insights into the secondary structure of amyloid proteins and their aggregates;
- Atomic force microscopy to obtain insights into aggregate morphology and physical parameters (height, length, width, periodicity);
- Mass spectrometry to determine mass of the protein;
- Mechanistic insights into amyloid aggregation process are obtained by exploiting bioinformatic tools such as rModeler or AmyloFit.